

In this blog post, I will show you guys how to calculate the kinetic energy density at the grid points and therefore the kinetic energy of the system. We will also compare the results with the analytically calculated kinetic energy.
In order to calculate the positive-definite kinetic energy density at a particular grid point, we will use the following formula [See Refs 1,2,3,4]:
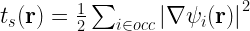
and then the total kinetic energy can be calculated using
which is numerically equivalent to
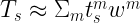
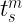
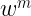
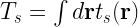
As you can notice from the first equation above, we require the knowledge of the molecular orbital coefficients of the occupied orbitals as well as the gradients of the atomic orbitals to evaluate the kinetic energy density. To get these quantities we can utilize the dft.mo_coeff attribute of a dft object and the dft.numint.eval_ao method with the deriv parameter equal to 1.
The python code to calculate the kinetic energy density at a grid point using PySCF is given below
Code
from pyscf import gto, dft
import numpy as np
# Create a variable that stores the x,y,z atomic coordinates
atomic_coordinates = '''
H 0.00000 0.00000 0.00000
H 0.74000 0.00000 0.00000
'''
mol = gto.Mole() # Create a Mole object
mol.atom = atomic_coordinates # Specify the coordinates
mol.basis = 'def2-SVP' # Specify the basis set
mol.build() # Build the Mole object
mf = dft.KS(mol) # Create a KS-DFT object and pass the mol object as argument
mf.xc = 'pbe' # shorthand for pbe,pbe [PBE exchange and PBE correlation]
mf.kernel() # Run the DFT calculation
# MO Coefficients of the occupied orbitals
occ_orbs = mf.mo_coeff[:, mf.mo_occ > 0.]
# Generate some grids (we could have also used the already generated grids though)
grids = dft.gen_grid.Grids(mol)
grids.build()
# Get the weights corresponding to the grid points
weights = grids.weights
# Get the first derivatives of the atomic orbitals at grid points
ao1 = dft.numint.eval_ao(mol, grids.coords, deriv=1, non0tab=grids.non0tab)
# The following gives the non-negative kinetic energy density at grid points
ts = 0.5 * np.einsum('xgp,pi,xgq,qi->g', ao1[1:,:,:], occ_orbs, ao1[1:,:,:], occ_orbs)
Ts = np.einsum('g,g->', weights, ts)
print('Numerical KE', Ts)
# Calculate KE analytically from the analytical kinetic potential matrix and contracting with the density matrix
Ts_ao = mol.intor('int1e_kin')
Ts_analyt = np.einsum('ui,uv,vi->', occ_orbs, Ts_ao, occ_orbs)
print('Analytical KE', Ts_analyt)
print('Diff b/w Analytical and Numerical Ts', np.abs(Ts-Ts_analyt))
You get the following output when you run the above program
Output
converged SCF energy = -1.1600212828208 Numerical KE 0.5503840418558041 Analytical KE 0.5503840487824062 Diff b/w Analytical and Numerical Ts 6.926602069690091e-09
References
- PhysRevB.91.035126 (aps.org)
- PhysRevB.75.155109 (aps.org)
- PhysRevB.91.045124 (aps.org)
- Semi-local machine-learned kinetic energy density functional demonstrating smooth potential energy curves – ScienceDirect
I’m a physicist specializing in computational material science with a PhD in Physics from Friedrich-Schiller University Jena, Germany. I write efficient codes for simulating light-matter interactions at atomic scales. I like to develop Physics, DFT, and Machine Learning related apps and software from time to time. Can code in most of the popular languages. I like to share my knowledge in Physics and applications using this Blog and a YouTube channel.