
The following is the documentation for CrysX: Crystallographic Tools v 1.1
For the documentations of CrysX-3D Viewer and CrysX-AR, visit https://www.bragitoff.com/crysx-3d-viewer-documentation/ and https://www.bragitoff.com/crysx-ar-documentation/ respectively.
Introduction
CrysX is a set of crystallographic tools available as apps / applications for Android devices. The tools are extremely handy for physics students, physicists and scientists working in the field of condensed matter physics. Computer softwares have long been used by crystallographers and condensed matter physicists to assist them in research. However, most of these softwares are only limited to traditional computer based operating systems such as Linux, Windows and MacOS. Nowadays, when smartphones have become an integral part of everyone’s life and their processing power easily matches that of home computers from a decade ago, it would be highly favorable to have such applications on tablets and mobile devices. Moreover, the easy-to-use interface along with high-quality tutorials makes using the apps really easy and makes a strong case for use as teaching aids in various courses such as solid state physics, nanoscience, etc.
CrysX is always under development and regularly gets updates with newer features and bug-fixes. In addition to the currently available tools and apps, newer tools are always being developed and will be available for download from this page.
Application Details
Download and Installation:
CrysX: Crystallographic Tools is available as a .apk file for the Android platform. The .apk file can be downloaded from here or installed directly from the Google Play Store. The advantage of installing it from the Google Play Store is that the app will get updated automatically. However, if you do download it from the former link, then you might have to enable a setting to ‘Allow apps from external sources‘ in your Android settings, to be able to install it successfully.
Features:
The app version 1.1 contains the Following Features:
- Powder X-Ray Diffraction Simulator
- Atomic Form Factor Calculator
- Interplanar Spacing Calculator
- Crystallite Size Calculator
- Space Group Decoder
- Custom CIF Creator
- CrysX – 3D Viewer (via an external app)
- CrysX – AR (via an external app)
- Equation of State Fitter
- Periodic Table
- Molar Mass Calculator
- Experimental XRD Pattern plotter from .xy file
- Gaussian Peak fitting of XRD pattern to calculate FWHM
Changes History v1.0:
- Powder X-Ray Diffraction Simulator
- Atomic Form Factor Calculator
- Interplanar Spacing Calculator
- Crystallite Size Calculator
- Space Group Decoder
- Custom CIF Creator
- CrysX – 3D Viewer (via an external app)
- CrysX – AR (via an external app)
- Equation of State Fitter
- Periodic Table
- Molar Mass Calculator
Report a Bug:
You can report a bug by either leaving a review on the Google Play Store, or leaving a comment here, or reaching out to the developer via email: [email protected]
Request a Feature:
You can request a feature by either leaving a review on the Google Play Store, or leaving a comment here, or reaching out to the developer via email: [email protected]
Usage Instructions, Tips and Examples
1. Powder Diffraction Simulator:
Powder diffraction simulator is the home page of ‘CrysX-Crystallographic Tools‘. You can use this to simulate the XRD pattern of any given crystal, provided its in the form of a .cif file. Compatible .CIF ifles can be downloaded from AMCSD or MaterialsProject.org. There are also some crystals provided in the application, so that you can test out the application for those, even if you don’t have .cif when you download the application. To use the provided CIF/crystals for simulation, just tap on the ‘Choose a category‘ drop-down menu and select a category, and the tap on ‘Select‘ to select a crystal structure belonging to the category. Once you select it, you see the simulated pattern within a few moments.
To use your own .CIF file from your phone storage, tap on the folder icon, and select a file from the storage. Once, selected the pattern will be simulated in a few moments.
Once the pattern is simulated it can be saved as an image by pressing the image icon above the simulated chart.
You can also change the wavelength for simulation, by entering the wavelength in angstroms, in the edit box on the right of ‘Enter the wavelength in Angstrom‘. The default is 1.5409 Angstrom.
The data corresponding to the peaks in the simulated pattern, such as hkl indices, positions, interplanar spacings, etc. can be exported as a .txt file by tapping on ‘Export data as .txt file‘, below the ‘Reflections‘ heading.
Watch the video below to see how can this module be used.
2. Atomic Form Factor Calculator
This, or any other module described in this documentation, can be used by tapping the hamburger menu button at the top-left of the screen in the title bar of the application. This will bring up a menu showing the various features/modules in the application.
To calculate the atomic form factor, select the atomic specie from the drop-down menu and the enter the value of the scattering vector between 0 and 25. Then press the ‘Go‘ button to calculate the corresponding atomic form factor.
Note: Regardless of the value of scattering vector entered, a chart of atomic form factor vs scattering vector is plotted as soon as the atomic species is selected.
3. Inter-planar Spacing Calculator
This can be used to calculate the inter-planar spacing from hkl Miller indices and lattice parameters, or from the peak position and wavelength of radiation used. One can also calculate the peak position from the inter-planar spacing using this module.
You just need to enter the required data in the edit-boxes and then press the ‘Submit‘ button for the corresponding calculations.
4. Crystallite Size Calculator
This module can be used to calculate the crystallite size using Scherrer formula. You need to provide the FWHM in degrees, wavelength in Angstrom, value of K (Scherrer constant) and the peak position in 2*theta (degrees) in the corresponding input fields and then press ‘Calculate‘ to get the result.
5. Space Group Decoder
The’ Space Group Decoder‘ allows you to enter a space group such as: ‘F D 3 M‘ and lists out the symmetry operations associated with it. Instead of space group name, one can also just enter the space group number, ex: 227.
6. Custom CIF Creator
As the name suggests, this module let’s the users enter the lattice parameters as well as the atomic positions in fractional coordinates and generates a .cif file. NOTE: Although, the module asks for the space group information, it doesn’t automatically generate the corresponding symmetrized atoms, nor does it lists the symmetry operations in the .CIF file. So you should ensure the you enter all the positions for the symmetric atoms as well.
7. Plot XRD Pattern
This module allows the users to plot the XRD pattern from a .xy file, whose first column contains the angle in 2*theta (degrees) and the second column contains the corresponding intensities. The plotted pattern can be saved as an image.
This module also allows the users to fit a Gaussian function to a peak to determine it’s FWHM for crystallite size calculation. To fit a peak, press the ‘Select the peak‘ button, and then zoom into the plotted chart and tap on the starting point of the peak, then press the ‘Confirm initial point‘ button. Then, tap on the ending point of the peak and press the ‘Confirm final point‘ buton. If you’re happy with the selected points and the region, press the ‘Fit‘ button to get the fitted parameters.
One can export the chart of the fitted peak, and use the FWHM in the ‘Crystallite Size Calculator‘ of the app to get the crystallite size.
8. CrysX-3D Viewer
This molecule/crystal visualizer and modeler has to be downloaded as an external app. It’s available for Windows, Linux and Mac OSX aas well. The visualizer enables the users to open popular .cif format files, to visualize the crystal structures of any compound. Even molecular structures can be visualized by opening popular .xyz format files. The visualizer is built using a gaming engine (Unity3d) ensuring stellar, never-before seen graphics on any other molecule/crystal visualizer. This makes the app really useful for researchers to prepare illustrations and figures for their research papers, thesis and dissertation. The app lets the users visualize lattice planes, and draw vectors to indicate electric/magnetic fields. Users can model supercells, monolayers (thin film/quantum well) or quantum dots. One can also edit the structures to create avacancy or introduce an impurity. There is also a feature that lets you draw your own custom 3D molecule/ nanocluster. Structures can also be analyzed by measuring the bond angles and lengths. Although, the app is quite straight-forward to use, high quality YouTube tutorials and documentation will get you up to speed in no time. If you find a bug or have any queries or want to request a feature you can post your question here: http://physwhiz.bragitoff.com with the tag ‘CrysX‘.
More details: https://www.bragitoff.com/crysx-3d-viewer/
9. CrysX-AR
CrysX – AR is an Android app that let’s you visualize molecules and crystals in Augmented Reality. The tool is powered by Google ARCore. The app can open .cif and .xyz files. This tool can help you make your poster presentations stand apart. Even classroom presentations can be made more intuitive.
More details: https://www.bragitoff.com/crysx-ar-documentation/
10. EOS Fitter: E(V)
You can either enter the data manually, or use a .TXT or .CSV file with the volume in cubic Angstroms in the first column and the energy in Rydbergs in the second column. This tool is especially useful for usage with Quantum ESPRESSO or VASP.
Sample Input file: vol_vs_E_Calcium_Bulk.txt, vol_vs_energy1.txt, vol_vs_energy2.txt, vol_vs_energy3.txt
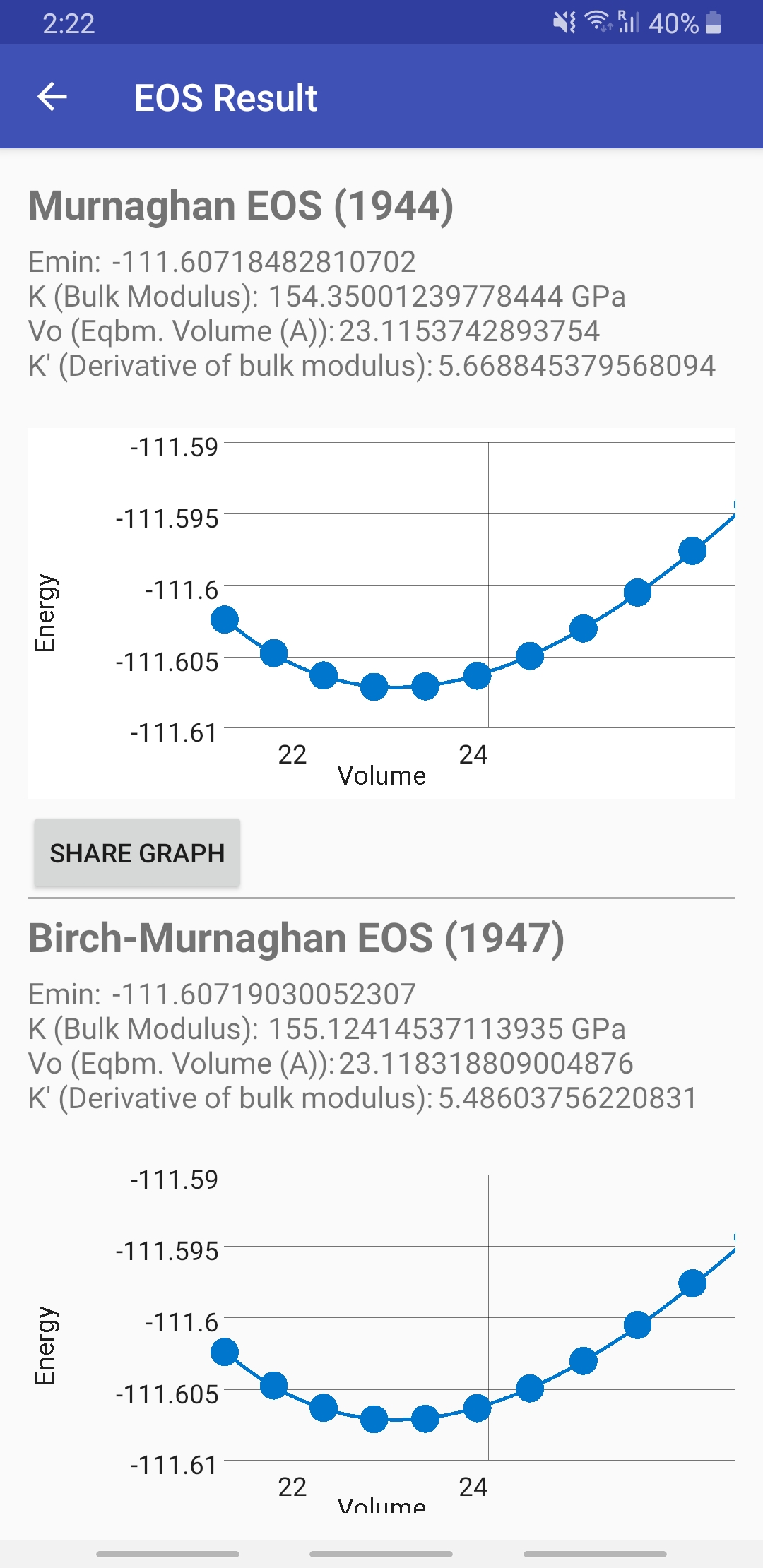
11. EOS Theory


12. File Conversions
This is a really useful module that let’s you interchange between various file formats and input files. Currently you can read .XYZ, .CIF, Quantum ESPRESSO input files, ORCA input files, etc. Once, read the structures can then be saved as all of the aforementioned formats. For example, if you read .XYZ file then the module will read the atomic positions, which can then be saved as ORCA input file. Moreover, the module will also assign a unit cell with lattice parameters large enough (with around 15 Angstrom of vacuum) to avoid periodicity effects. Therefore, if you save the structure as .CIF or QE input file then it will also have this lattice information. Overall, this module can be used to see if the QE/ORCA input file you created is correct or not by letting you convert the files into a .CIF/.XYZ and then visualizing them either in CrysX-3D Viewer or any external visualizer.
NOTE: Since there are a lot of conventions regarding the input file creations for Quantum ESPRESSO, and various existing formats and standards for .CIF, this process may not always work as expected. The module also doesn’t support the use of celldm() in QE input files. The lattice parameters should be provided using A, B, C, cosab, etc. CELL_PARAMETERS is also not supported. Users may also face some problems with some complicated lattices ex: ibrav=-3, ibrav=-13, etc. In our tests Quantum ESPRESSO input files created BURAI seemed to be best supported by this module.
13. Periodic Table
This module can be used to get chemical properties of the elements of the periodic table. One can tap on any element o get the various properties such as, atomic mass, number, symbol, electronic configuration, electronegativity, atomic radius, ionic radius, van der Walls radius, covalent radius, ionization energies, etc. Alternatively, users can tap on the search icon on the upper-right corner in the title bar to look for an element.
14. Molar Mass Calculator
This module let’s the users calculate the molar mass of any chemical compound. One just needs to enter the compound’s chemical formula into the input field, ex: CH3COOH, or CO2, or H2O.
Note: Parentheses are not supported yet.
15. GIF Creator
This module let’s users select a set of images and import them as .GIF with a frame rate of 2 FPS and desired resolution. NOTE: Higher resolutions and higher number of images take longer times.
This can be useful to create animations showing the structure movements or the molecular dynamics. All one needs to do is take the necessary screenshots with CrysX-3D Viewer and then use this module to stitch them into a GIF.
16. Math Tools
This Math toolbox has to be downloaded as an external application, and can be used to perform various matrix operations, plotting, curve-fitting, equation solving, root finding, numerical calculus, etc.
More details: https://play.google.com/store/apps/details?id=com.bragitoff.themathapp&hl=en
YouTube Tutorials:
Open Source Libraries
The app uses the following open source libraries, and the developer thanks the awesome community:
For Android App:
- Fancy Buttons by Mehdi Sakout
- Android About Page by Mehdi Sakout
- mXparser
- Apache Commons Math Library
- Material File Picker by nbsp-team
- GraphView by jjoe64
- MP Android Chart by Phil Jay
- android-ndk-gif by waynejo
- MultiType-FilePicker by fishwjy
For PC/MacOS/Linux Application:
Special Notes
It should be noted, that due to the large variety of .CIF formats out there, CrysX may not yet support all of them as the application is still in the budding stage. However, if you ever encounter a file that fails to be parsed, let me know and mail the file to me. I will try to add the compatibility for the various formats. Similarly while file conversion, for some complicated systems the converted CIF or QE Input may not be what you expected. Please let me know if you encounter a problem.
Legal Disclaimer
While I try to make everything as accurate as possible, with honesty, the following disclaimer applies to all the softwares I create:
This software and any accompanying documentation are released “as is.” I make no warranty of any kind, expressed or implied, concerning this software and any accompanying documentation, including without limitation, any warranties of merchantability or fitness for a particular purpose. In no event will I be liable for any damages, including any lost profits, lost savings, or other incidental or consequential damages arising out of the use, or inability of use, of this software or any accompanying documentation, even if informed in advance of the possibility of such damages.
Users are advised to make sure and confirm that the results obtained through CrysX: Crystallographic Tools are correct or not on their own and I cannot be held responsible for faulty results. Users are responsible for their results, not me.
There is absolutely no WARRANTY associated or provided with the use of this software. This program is distributed in the hope that it will be useful, but without any warranty; without even the implied warranty of merchantability or fitness for a particular purpose.
License
CrysX: Crystallographic Tools is currently copyrighted with its source code not open to the public; we wish to control the development and future of CrysX: Crystallographic Tools by ourselves. This software is distributed free of charge for academic, scientific, educational, and commercial users. Permission to use this software is hereby granted under the following conditions:
- Results or calculations done/ produced by CrysX: Crystallographic Tools may be used in any publications (original and review articles) provided that its use is explicitly acknowledged. A suitable reference for CrysX: Crystallographic Tools is:
Sharma, M. & Mishra, D. (2019). J. Appl. Cryst. 52, 1449-1454.
Frequently Asked Question about CrysX: Crystallographic Tools
Can I export the simulated pattern as image?
Yes!
Can I export the data of the simulated pattern?
Yes!
Can I export the image of the fitted peaks?
Yes!
Can I export the EOS (Equation of State) curves?
Yes!