

The Kohn-Sham approach in density functional theory (DFT) is a true game-changer in tackling the intricate many-body problems of quantum mechanics. By introducing an auxiliary system of non-interacting particles, this approach reduces the complex many-body problem to a tractable set of single-particle equations. Moreover, by explicitly separating the kinetic energy of independent particles and the long-range Hartree term, the remaining unknown quantity – the exchange-correlation functional ![E_{XC}[\rho]](https://s0.wp.com/latex.php?latex=E_%7BXC%7D%5B%5Crho%5D&bg=ffffff&fg=000&s=0&c=20201002)
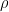

Despite the exact
The Local Density Approximation (LDA)
The local density approximation (LDA) forms the foundation of all approximate exchange-correlation functionals. At its core lies the concept of a uniform electron gas – a system where electrons move in a positive background charge distribution, ensuring overall charge neutrality.
In LDA, the exchange-correlation energy is expressed as:
![E_{XC}^{LDA}[\rho] = \int \rho(\vec{r}) \epsilon_{XC}(\rho(\vec{r})) d\vec{r}](https://s0.wp.com/latex.php?latex=E_%7BXC%7D%5E%7BLDA%7D%5B%5Crho%5D+%3D+%5Cint+%5Crho%28%5Cvec%7Br%7D%29+%5Cepsilon_%7BXC%7D%28%5Crho%28%5Cvec%7Br%7D%29%29+d%5Cvec%7Br%7D+&bg=ffffff&fg=000&s=2&c=20201002)
Here, 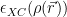

The exchange part, 

However, no such closed-form expression exists for the correlation part, 
The LDA can be extended to the local spin-density approximation (LSDA) by employing the two spin densities, 

While LDA provides a good starting point, its accuracy is often insufficient for chemistry applications, with errors of 10-20% in quantities like ionization energies, dissociation energies, and cohesive energies. However, it surprisingly yields bond lengths with an accuracy of ~2%.
The Generalized Gradient Approximation (GGA)
The next step beyond LDA is the generalized gradient approximation (GGA), which incorporates the magnitude of the density gradient 
![E_{XC}^{GGA}[\rho_\uparrow, \rho_\downarrow] = \int f(\rho_\uparrow, \rho_\downarrow, \nabla\rho_\uparrow, \nabla\rho_\downarrow) d\vec{r}](https://s0.wp.com/latex.php?latex=E_%7BXC%7D%5E%7BGGA%7D%5B%5Crho_%5Cuparrow%2C+%5Crho_%5Cdownarrow%5D+%3D+%5Cint+f%28%5Crho_%5Cuparrow%2C+%5Crho_%5Cdownarrow%2C+%5Cnabla%5Crho_%5Cuparrow%2C+%5Cnabla%5Crho_%5Cdownarrow%29+d%5Cvec%7Br%7D+&bg=ffffff&fg=000&s=2&c=20201002)
GGAs are often split into separate exchange and correlation contributions, with the exchange part taking the form:
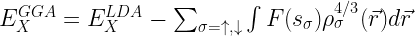
where 
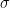
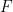

The correlation counterparts are even more complex, with widely used options like Perdew’s P86, Perdew and Wang’s PW91, and Lee-Yang-Parr (LYP) functionals.
Commonly used GGA combinations include BP86 (Becke exchange with P86 correlation), BLYP (Becke exchange with LYP correlation), and BPW91 (Becke exchange with PW91 correlation).
GGAs have reduced the LDA errors in atomization energies of small molecules by a factor of 3-5, making DFT a widely adopted tool in quantum chemistry.
Hybrid Functionals
Accurate exchange functionals are crucial for meaningful DFT results, as exchange contributions often dominate over correlation effects. Hybrid functionals leverage the fact that the exact exchange energy of a Slater determinant can be computed using the Hartree-Fock (HF) method.
These functionals combine the orbital-dependent HF exchange with an explicit density functional. One popular approach, introduced by Becke, approximates the total exchange-correlation energy as:

where DFA denotes an LDA or GGA functional.
The widely used B3LYP functional, proposed by Becke, Lee, Yang, and Parr, defines the exchange-correlation energy as:

where the coefficients 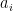
Hybrid functionals like B3LYP are the method of choice in the quantum chemistry community due to their accuracy in predicting energetics.
In conclusion, while the exact exchange-correlation functional remains elusive, the development of approximations like LDA, GGA, and hybrid functionals has been instrumental in propelling density functional theory to the forefront of computational chemistry and physics. As researchers continue to refine and develop new functionals, the predictive power and applicability of DFT will only continue to grow.
References
- Exchange-correlation Functionals | Johannes Voss (stanford.edu)
- Density functional theory – Wikipedia
- Hybrid functional – Wikipedia
- Local-density approximation – Wikipedia
- Molecular Electronics | World Scientific Series in Nanoscience and Nanotechnology
- 2303.11370 (arxiv.org)
- 1062vch06.p65 (ucsb.edu)
I’m a physicist specializing in computational material science with a PhD in Physics from Friedrich-Schiller University Jena, Germany. I write efficient codes for simulating light-matter interactions at atomic scales. I like to develop Physics, DFT, and Machine Learning related apps and software from time to time. Can code in most of the popular languages. I like to share my knowledge in Physics and applications using this Blog and a YouTube channel.