


In Kohn-Sham density functional theory (KS-DFT), the ground state electron density is described using a single mathematical determinant, which leads to a set of equations that are similar to those used in Hartree-Fock theory and are known as Kohn-Sham equations:
![\left[-\frac{\hbar^2}{2m}\nabla^2 + v_{\text{ext}}(\mathbf{r}) + v_{\text{H}}(\mathbf{r}) + v_{\text{xc}}(\mathbf{r})\right] \psi_i(\mathbf{r}) = \epsilon_i \psi_i(\mathbf{r})](https://s0.wp.com/latex.php?latex=%5Cleft%5B-%5Cfrac%7B%5Chbar%5E2%7D%7B2m%7D%5Cnabla%5E2+%2B+v_%7B%5Ctext%7Bext%7D%7D%28%5Cmathbf%7Br%7D%29+%2B+v_%7B%5Ctext%7BH%7D%7D%28%5Cmathbf%7Br%7D%29+%2B+v_%7B%5Ctext%7Bxc%7D%7D%28%5Cmathbf%7Br%7D%29%5Cright%5D+%5Cpsi_i%28%5Cmathbf%7Br%7D%29+%3D+%5Cepsilon_i+%5Cpsi_i%28%5Cmathbf%7Br%7D%29+&bg=ffffff&fg=000&s=1&c=20201002)
where 

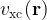
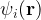

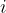
The total energy is composed of the non-interacting kinetic energy 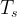
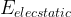
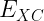
![T_c[\rho] = T[\rho]-T_s[\rho]](https://s0.wp.com/latex.php?latex=T_c%5B%5Crho%5D+%3D+T%5B%5Crho%5D-T_s%5B%5Crho%5D+&bg=ffffff&fg=000&s=1&c=20201002)
![E[\rho] = T_s + E_{elecstatic}[\rho] + E_{XC}[\rho]](https://s0.wp.com/latex.php?latex=E%5B%5Crho%5D+%3D+T_s+%2B+E_%7Belecstatic%7D%5B%5Crho%5D+%2B+E_%7BXC%7D%5B%5Crho%5D+&bg=ffffff&fg=000&s=1&c=20201002)
Approximations
Here are some of the main approximations used in Kohn-Sham DFT:
-
- In KS-DFT, one avoids the need for a kinetic energy functional
by evaluating the exact kinetic energy
from the wavefunction (single Slater determinant) of a fictitious non-interacting system with the same density as that of the real one. Therefore, KS-DFT calculates the kinetic energy in terms of orbitals. However, one still needs to approximate the
(mentioned above). Since we are again working with orbitals in KS-DFT (like in the case of Hartree-Fock theory), we need a set of basis functions (atomic orbitals) for each atom of our system whose linear combination corresponds to KS orbitals (molecular orbitals). This set of basis functions is called a basis set and makes the calculation accuracy dependent on the quality of basis sets used.
- Use of the exchange-correlation functional, which approximates the exchange and correlation energy of a many-electron system. The exchange-correlation energy accounts for the remaining electronic energy not included in the non-interacting kinetic and electrostatic terms. Some of the common types are: local (LDA), semi-local (GGA), and hybrid (including some portion of exact exchange) functionals that differ in accuracy.
- Approximate exchange-correlation functionals also lead to self-interaction errors (SEI) which in turn lead to delocalization errors. In both HF theory and KS-DFT, the potential field includes the Coulomb potential, which is the interaction of the electron with the entire electron density of the atom, molecule, or material. That is physically incorrect because an electron does not interact with itself. In HF theory, however, the exchange potential (non-local in nature) completely cancels the self-interaction part of the Coulomb potential which is not the case for KS-DFT, due to the approximate (not completely non-local) exchange-correlation functionals. It has been known to be the primary cause for the underestimation of band-gaps in semiconductors.
- Ignoring relativistic effects, which can be important for certain types of systems. Standard DFT does not take into account relativistic effects like spin-orbit coupling. However, many codes have the ability to account for it by using relativistic pseudopotentials.
- Use of Born-Oppenheimer approximation. The positions of the nuclear cores are considered parameters of the electronic Hamiltonian thus defining a potential energy surface. The electronic system is assumed to adiabatically follow changes in the nuclear coordinates relaxing to its ground state on much faster timescales than the timescales of nuclear motion (electron-phonon coupling, e.g., is a correction to this adiabatic approximation).
- In KS-DFT, one avoids the need for a kinetic energy functional
If you have any questions/doubts or find any mistake in the above article then please let me know in the comments section down below.
References:
- density functional theory – What correlation effects are included in KS-DFT with LDA and GGA? – Matter Modeling Stack Exchange
- Perspective: Kohn-Sham density functional theory descending a staircase: The Journal of Chemical Physics: Vol 145, No 13 (scitation.org)
- Status and Challenges of Density Functional Theory (umn.edu)
- Self-Interaction Error in Density Functional Theory: An Appraisal | The Journal of Physical Chemistry Letters (acs.org)
- computational chemistry – Striking examples where Kohn-Sham orbitals clearly have no physical meaning – Chemistry Stack Exchange
- What Do the Kohn−Sham Orbitals and Eigenvalues Mean? | Journal of the American Chemical Society (acs.org)
- Slide 1 (southampton.ac.uk)
- Pantazis_DFT-BasisSets (uni-paderborn.de)
- basis-sets.pdf (gatech.edu)
- density functional theory – Spin–orbit interaction with DFT – Matter Modeling Stack Exchange
- Self-interaction corrections in density functional theory: The Journal of Chemical Physics: Vol 140, No 18 (scitation.org)
Other useful resources:
I’m a physicist specializing in computational material science with a PhD in Physics from Friedrich-Schiller University Jena, Germany. I write efficient codes for simulating light-matter interactions at atomic scales. I like to develop Physics, DFT, and Machine Learning related apps and software from time to time. Can code in most of the popular languages. I like to share my knowledge in Physics and applications using this Blog and a YouTube channel.