
Crystal Structure:
| Co (Hexagonal Closed Pack) |
|
About/Help |
CIF Source:
Taylor A, Floyd R W
Acta Crystallographica 3 (1950) 285-289
Precision measurements of lattice parameters of non-cubic crystals
Locality: synthetic
Sample: at T < 450 C
_database_code_amcsd 0009140
http://rruff.geo.arizona.edu/AMS/download.php?id=10374.cif&down=cif
Simulated Powder XRD using VESTA:
X-Ray Wavelength: 1.54059 Angstrom
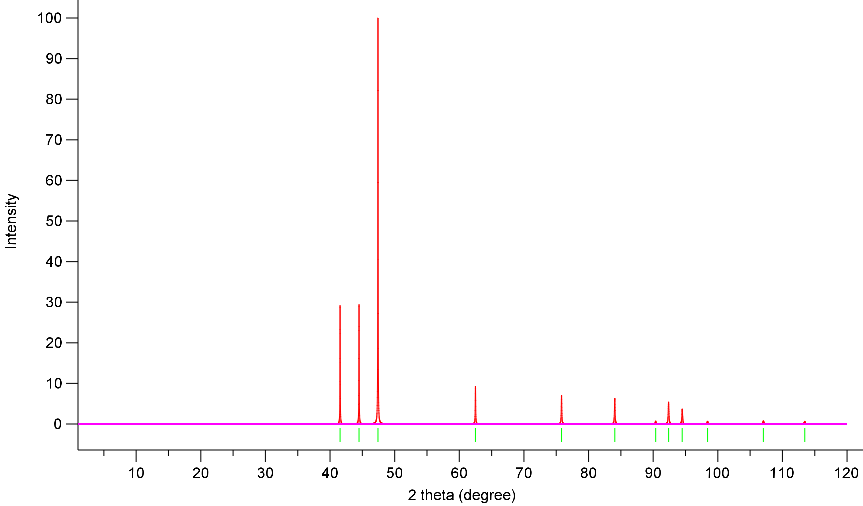
Simulation 1: GGA-Spin Polarized
Pseudopotential Used:
Co.pbe-n-rrkjus_psl.1.0.0.UPF
PP Type: Ultrasoft
Exchange Correlation Functional: PBE-GGA Spin Polarized
Non-linear core corrections are used.
Wavefunction Energy Cutoff: 47 Ry
Charge Density Energy Cutoff: 517 Ry
k – mesh: 8x8x8
Run Type: GGA-PBE Spin Polarized
Starting Magnetization: 0.8
Total Energy vs Cutoff:
Cutoff(Ry) Total Energy(Ry)
25 -152.10091494
30 -152.25971773
35 -152.27675576
40 -152.27775041
45 -152.28072344
47 -152.28215106
50 -152.28389648
In order to perform spin polarized calculations set the nspin parameter to 2.
Then as explained here, set a starting magnetization to break the symmetry. The calculation should find the lowest-energy spin state compatible with the given crystal structure and not orthogonal to initial conditions (e.g.: if you start
with a FM alignment, you will hardly find an AFM final state even if it exists). Perform several calculations at different starting magnetizations, choose the one with smaller energy as ground state. The system must be in all cases treated as a metal, whether it is or not. In principle, you should use pseudopotentials with the nonlinear core correction.
The following shows the total energy for different values of starting magnetization. NOTE: Starting magnetization is given in fractions, ranging between -1 (all spins down for the valence electrons of atom type ‘i’) to 1 (all spins up).
Total Energy vs Starting Magnetization:
SM Total Energy (Ry) Tot. Magnetic Mom/Abs. Mg. Mom. (Bohr Magneton)
0.1 -152.24120855 0/0
0.2 -152.28215106 3.28/3.64
0.3 -152.28215155 3.28/3.64
0.4 -152.28215149 3.28/3.64
0.5 -152.28215153 3.28/6.64
0.6 -152.28215146 3.28/3.64
0.7 -152.28215147 3.28/3.64
0.8 -152.28215157 3.28/3.64
0.9 -152.28215131 3.28/3.64
1.0 -152.28215119 3.28/3.65
Clearly, a starting magnetization value of 0.8 gives the lowest energy.
Now, we perform optimization of geometry.
Optimized Coordinates and Lattice Parameters:
CELL_PARAMETERS {angstrom}
2.491858 0.000001 -0.000000
-1.245928 2.158011 0.000000
-0.000000 0.000000 4.014676
ATOMIC_POSITIONS {angstrom}
Co -0.000009 1.438683 1.003669
Co 1.245927 0.719351 3.011007
Total magnetic moment for optimized system: 3.25 Bohr Magneton.
Since there are 2 Co atoms in our HCP lattice, therefore, the total magnetization per atom is 3.25/2=1.625 Bohr. Magnt. which is astoundingly very close to the experimental value.
Magnetic moment per atom= 1.625 B.M.
Bandstructure:

Density of States(DOS):
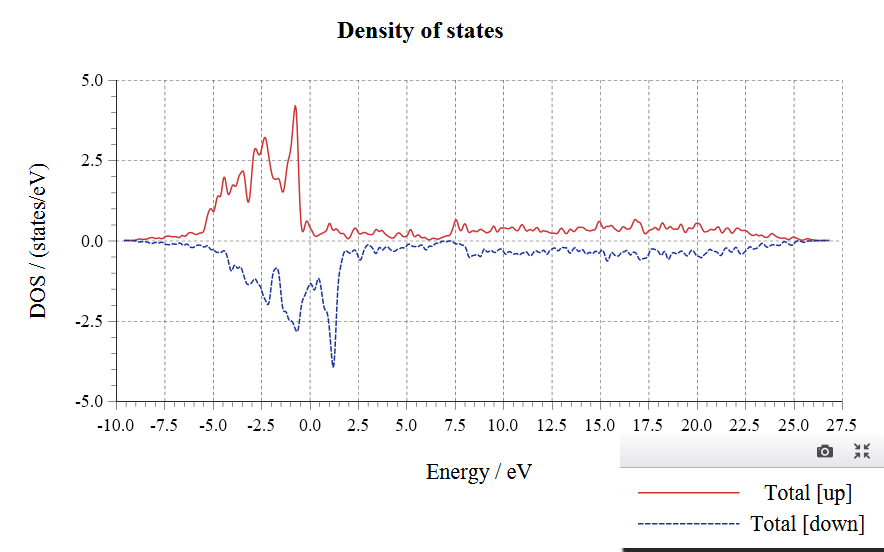
Input Files:
Acknowledgements:
I acknowledge the use of the following tools and packages in order to produce the above simulations.
Quantum Espresso(for DFT based simulations): http://www.quantum-espresso.org/
BURAI(for visualization and as a GUI for QE): http://nisihara.wixsite.com/burai
VESTA(for visualization and XRD simulations): http://jp-minerals.org/vesta/en/
References and Resources
https://en.wikipedia.org/wiki/Cobalt
http://www.materialsdesign.com/appnote/magnetic-moment-iron
http://www-rjn.physics.ox.ac.uk/lectures/magnetismnotes10.pdf
http://146.141.41.27/Lectures/Omololu-Wednesday-21-MetalsMagnetism2.pdf
I’m a physicist specializing in computational material science with a PhD in Physics from Friedrich-Schiller University Jena, Germany. I write efficient codes for simulating light-matter interactions at atomic scales. I like to develop Physics, DFT, and Machine Learning related apps and software from time to time. Can code in most of the popular languages. I like to share my knowledge in Physics and applications using this Blog and a YouTube channel.