
The following is a DFT based study of Silicon (Diamond FCC) crystal using the package Quantum Espresso.
Crystal Structure:
| Silicon |
|
About/Help |
CIF Source:
Wyckoff R W G
Crystal Structures 1 (1963) 7-83
Second edition. Interscience Publishers, New York, New York
_database_code_amcsd 0011243
http://rruff.geo.arizona.edu/AMS/CIF_text_files/13231_cif.txt
Simulated Powder XRD using VESTA:
X-Ray Wavelength: 1.54059 Angstrom
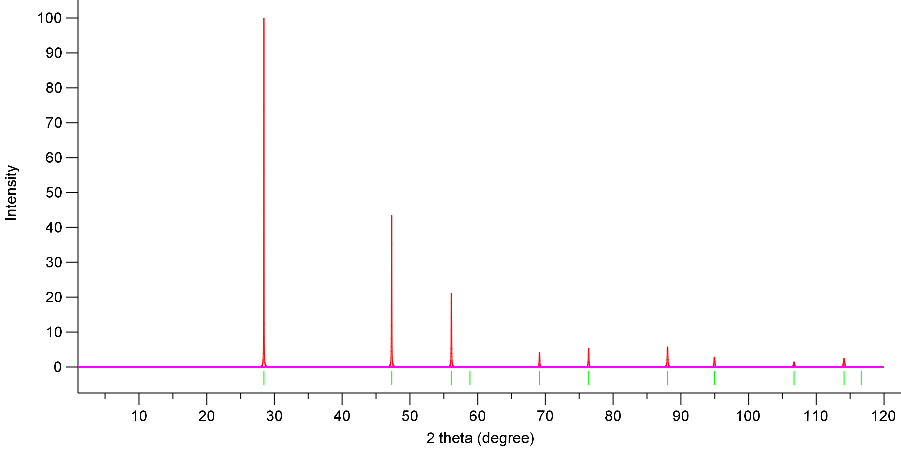
Simulation 1: GGA
Pseudopotential Used:
Si.pbe-nl-rrkjus_psl.1.0.0.UPF
P.P. Type: Ultrasoft
Exchange Correlation Functional: PBE-GGA
Non-linear Core Corrections Used
Total Energy vs Cutoff:
Cutoff(Ry) Total Energy(Ry)
15 -81.72897323
20 -81.74078101
25 -81.74871306
30 -81.75301878
35 -81.75465390
40 -81.75556513
45 -81.75597750
50 -81.75612289
55 -81.75617006
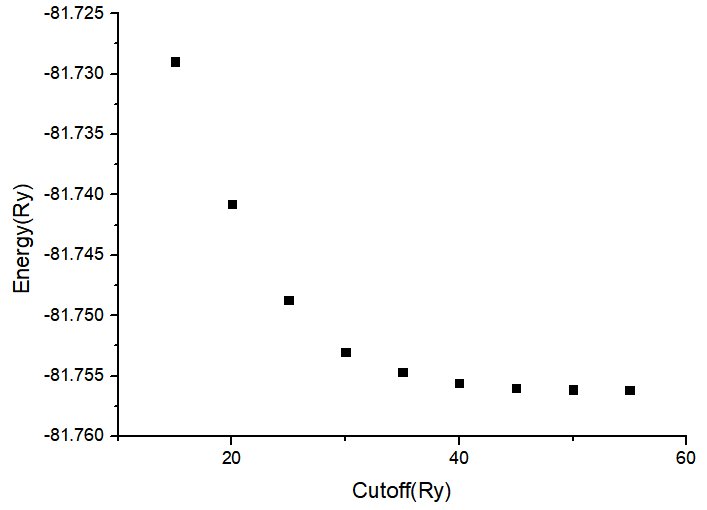
Wavefunction Energy Cutoff: 45 Ry
Charge Density Energy Cutoff: 450 Ry
k – mesh: 12x12x12
Run Type: GGA-PBE
Optimized Coordinates and Lattice Parameters:
Lattice Parameters: a=b=c= 5.46735 A, alpha=beta=gamma=90 deg
ATOMIC_POSITIONS {angstrom}
Si 0.000000 0.000000 0.000000
Si 0.000000 2.733677 2.733677
Si 2.733677 0.000000 2.733677
Si 2.733677 2.733677 0.000000
Si 4.100516 4.100516 1.366838
Si 4.100516 1.366838 4.100516
Si 1.366838 4.100516 4.100516
Si 1.366838 1.366838 1.366838
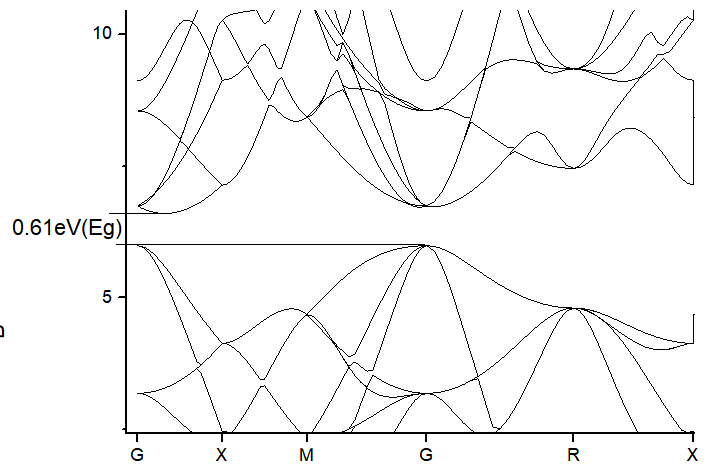
Bandstructure:
Band-Gap: 0.61 eV
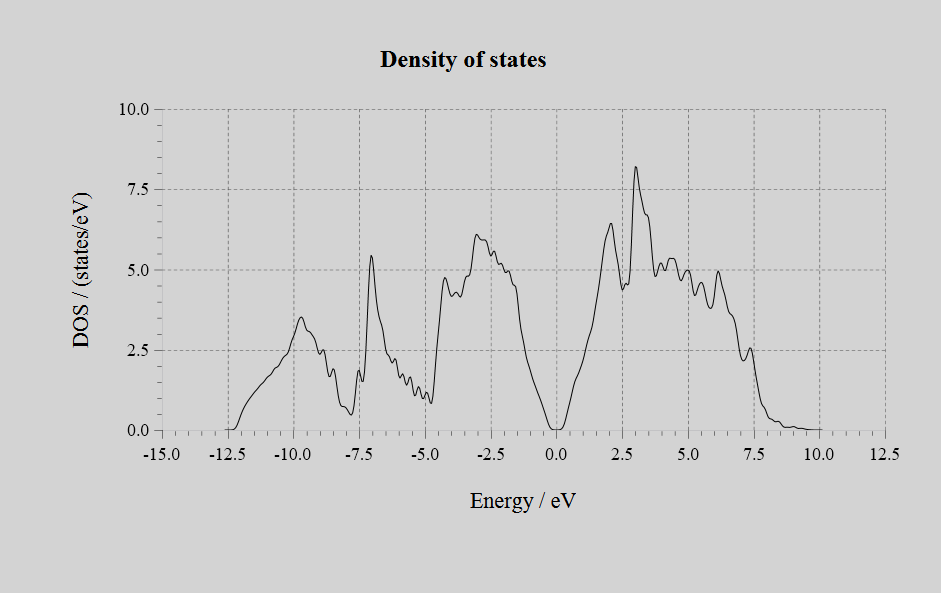
Density of States(DOS):
Input Files:
Silicon_QuantumEspressoInputFiles_DFT
Simulation 2:(HSE – Hybrid Functional)
Screening Parameter: 0.0916
q-grid: 8x8x8
k-grid: 8x8x8
DFT Type: HSE
highest occupied, lowest unoccupied level (ev): 6.0340 7.2744
Band Gap: 1.2404 eV
Input files:
Acknowledgements:
I acknowledge the use of the following tools and packages in order to produce the above simulations.
Quantum Espresso(for DFT based simulations): http://www.quantum-espresso.org/
BURAI(for visualization and as a GUI for QE): http://nisihara.wixsite.com/burai
VESTA(for visualization and XRD simulations): http://jp-minerals.org/vesta/en/
I’m a physicist specializing in computational material science with a PhD in Physics from Friedrich-Schiller University Jena, Germany. I write efficient codes for simulating light-matter interactions at atomic scales. I like to develop Physics, DFT, and Machine Learning related apps and software from time to time. Can code in most of the popular languages. I like to share my knowledge in Physics and applications using this Blog and a YouTube channel.
Hai, I am currently working as Research Associate,…. recently, i started working on condensed matter theory.. for which i am learning….DFT calculation. I have some experience in Quantum expresso.
I want to know about ‘Burai GUI of QE’
I have Pearson Crystal Database software.
In Burai, I am not able to open .cif files downloaded from Pearson Crystal Database software.
Can you please give me the solution for this………
Hi there,
I haven’t used the Pearson Crystal Database, so I can’t tell you what the problem might be. Maybe you could send me a copy of the .cif file at [email protected] and I’ll take a llok.
Also, if you have the .cif file, then creating a Quantum Espresso input file shouldn’t be too tough. You could try to create a QE input file and open that using BURAI.
why you are getting silicon bandgap 0.61 eV instead 1.1 eV (at 300k)
Because of the well known problem with traditional DFT-GGA: the over-delocalization of the VB electron states
i think burai1.3 with worked based upon LDA approximation?? Am I right?
Second how to get rid it.I think there is an option named GGA+U approximation but it required hubbard parameter.what is the value of hubbard parameter for silicon.
Actually hubbard correction is not very suitable solution for Silicon. You should go with HSE functionals. They give good results for Si.
Also the functinal is decided by the pseudopotential. So if you’re using a PBE functional then it isa type of GGA
Hi Manas, you did great job, but I didint get about “Screening Parameter: 0.0916”, how this value achieved. please help me sir.